Longitudinal multi-omics analysis reveals subset-specific mechanisms underlying irritable bowel syndrome
Commented articles
By Prof. Harry Sokol
Gastroenterology and Nutrition Department, Saint-Antoine Hospital, Paris, France
Lay public section
Find here your dedicated section
About this article
Author

Created
12 August 2021
Updated
12 August 2024
Commentary on the original article by Mars et al. Cell 2020 [1]
The gut microbiome is implicated in numerous chronic gastro-int estinal disorders in humans. Determining its role however has been rendered difficult due to the lack of correlation between animal and human studies as well as that of an integrated multi-omics view of disease-specific physiological changes. The authors integrated longitudinal multi-omics data from the intestinal microbiome, the metabolome, the host epigenome and the transcriptome in the context of irritable bowel syndrome (IBS) host physiology. They have identified IBS sub-type specific and symptom-related variations in microbial composition and function. A sub-group of changes identified in microbial metabolites corre sponds to host physiological mechanisms which are relevant to IBS. By compiling multiple data layers, the authors identified purine metabolism to be a new host-micro biota metabolic pathway in IBS with potential for therapeutic application. This study highlights the value of longitudinal sampling and the integration of complementary multi-omics data in the identification of functional mechanisms which may be future therapeutic targets in a global treatment strategy for chronic intestinal diseases.
WHAT DO WE ALREADY KNOW ABOUT THIS SUBJECT?
Irritable bowel syndrome (IBS) is a disorder observed in patients all over the world, characterised by recurrent abdominal pain or discomfort. Occurring mainly in women, IBS is associated with changes in the form or frequency of stools and it is the form of the latter which defines the IBS subsets: IBS with constipation (IBS-C), IBS with diarrhoea (IBS-D) or IBS with mixed bowel habits (IBS-M).
The pathogenesis of IBS involves changes in gastro-intestinal motility, intestinal secretion, visceral hypersensitivity and intestinal permeability, all of which can be modified by the gut microbiome [2]. Moreover, IBS symptoms are influenced by the diet, host genetics and the environment, which are also known to modulate the human gut microbiome [2]. Experimental evidence supporting the role of the gut microbiome in IBS are based on patient-to-gnotobiotic mouse transplantation studies which reproduced certain symptoms associated with IBS-C and IBS-D (transit time, sensation of pain, intestinal permeability…) [3]. However, in the absence of robust animal models of IBS, studies in humans are needed to determine the interactions between the gut microbiome and the pathological pathways specific to humans. Human IBS studies are limited in general by the use of transversal sampling and a failure to stratify into patient subsets, which is reflected in the lack of agreement in the results obtained in the large number of studies on the microbiome [4].
The well-described influence of gastro-intestinal transit on the intestinal microbiome increases the variability of the studies even more. In addition, IBS, in common with other chronic gastro-intestinal disorders, is characterised by periods of remission and flare up of the symptoms, and transversal sampling therefore fails to take into account the fluctuations of the disease over time. Lastly, the inherent differences in host physiology between humans and animals have been an obstacle to progress in our understanding of the mechanistic roles of the gut microbiome in IBS. The authors performed a longitudinal study in subsets of patients with IBS, integrating multi-omics measurements, including the microbial metagenome, host transcriptome and the methylome with assessment of host cell functions. This revealed mechanisms specific to the IBS subset induced by impaired microbial metabolism, which corresponded to simultaneous changes in the host physiology.
Image
WHAT ARE THE MAIN INSIGHTS FROM THIS STUDY?
Here, the authors conducted a prospective observational longitudinal study with multi-omics analysis of the gut microbiome and the host cells. Healthy subjects were compared with patients with IBS-C and IBS-D. A total of 77 participants supplied at least one stool sample (a total of 474 stool samples were obtained), and 42 participants received a sigmoidoscopy which provided colon biopsies. In order to identify the microbial factors causing the symptoms specific to each of the IBS subsets, metagenomic sequencing and a metabolomic analysis were performed on the stool samples. A metabolomic analysis and cytokine assays were performed on serum samples. Lastly, 16S sequencing, and metabolomic, transcriptomic and methylomic analyses were performed on the colon biopsies. The authors identified differences in the composition and diversity of the gut microbiota between healthy subjects and IBS-C or IBS-D patients.
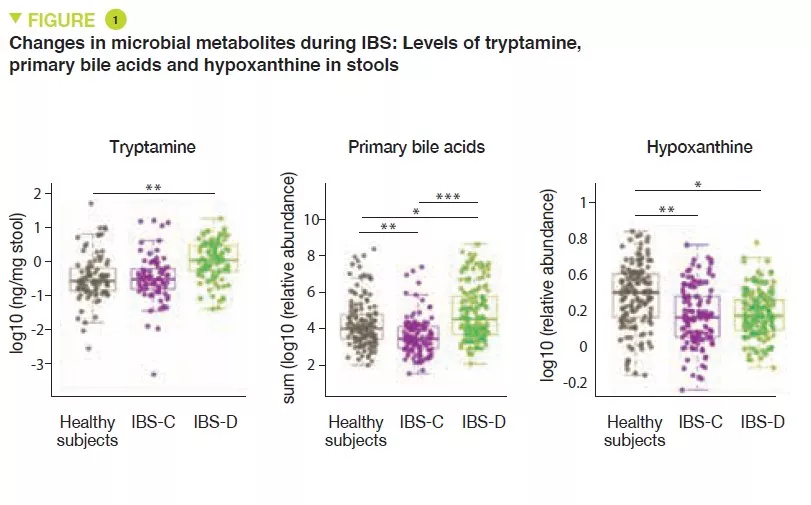
Metabolomic analysis of the stools revealed increased levels of tryptamine, a tryptophan metabolite produced by certain intestinal bacteria, in patients with IBS-D (Figure 1). Because tryptamine accelerates transit due to an action on the serotonin receptor, 5-HT4, it is suggested that it could have a role in the phenotype of these patients. Similarly, the proportion of primary bile acids was higher in patients with IBS-D, a sign of impaired transformation of bile acids by the microbiota. In vitro experiments suggest that primary bile acids increase secretion in the colon and could also participate in the phenotype.
Lastly, the integration of multi-omics data identified a potential new mechanism in IBS. The results suggest that in IBS patients there is increased degradation of purine nucleotides and of hypoxanthine in particular, by the microbiota and the host cells, which induces stress in the colon. It is suggested that this could lead to a compensatory response with an increase in purine recuperation. The low levels of purine nucleotides could lead to a reduced source of energy to the mucosal epithelium and to a reduced capacity for repair of the gut mucosa, which could partly contribute to IBS physiopathology.
WHAT ARE THE CONSEQUENCES IN PRACTICE?
These data suggest a role of the gut microbiota in the physiopathology of IBS with differences between IBS-C and IBS-D. Moreover, these results point towards the potential role of a deficiency in purine nucleotides, in particular via an over-consumption of hypoxanthine by the microbiota and the host cells. These results point the way to treatments stimulating the production of microbial hypoxanthine or inhibiting xanthine oxidase locally in the intestine.
Conclusion
This longitudinal study with integrated multi-omics shows the value of longitudinal studies in humans and highlights functional alterations to the microbiota in IBS which may potentially be implicated in the physiopathology of this disease. The new leads which have been identified may be new therapeutic targets.
Sources