
Gut microbiome and antimicrobial resistance
By Dr. Joseph Nesme
Assistant professor, Section of Microbiology, Department of Biology, University of Copenhagen, Denmark
Lay public section
Find here your dedicated section
About this article
Created
17 October 2023
Updated
01 December 2025
Antibiotic resistance is ancient and prevalent and the environmental resistome is a reservoir of resistance genes fueling the antimicrobial resistance silent pandemic. Horizontal gene transfer, population dynamics and co-selection accelerate emergence and dissemination of the resistome and by year 2050, more than 10 million death per year will be due to antibiotic resistance. The early colonization of the gut microbiome is prone to dysbiosis that can lead to increased abundance and diversity of the gut resistome of infants. Adopting a One Health approach, considering the interconnectedness of human health, animal health, and the environment, is essential for addressing the complex challenges posed by AR.
Antibiotic resistance in the environment: A pre-existing challenge
Antibiotic resistance (AR) is an ancient and prevalent phenomenon in the environment existing long before the introduction of antibiotic molecules as therapeutic. The environment serves as a vast reservoir of antibiotic resistance genes with diverse microbial communities harboring resistance mechanisms. AR has been found in various environmental settings, including soil, water, plants, animals, and even in 30,000 years old Arctic permafrost 1, 2 . The ecological role of antibiotic molecules and associated resistance in non-clinical settings remains unclear but highlights the fact that a readily available pool of genes predates clinical antibiotic usage and explain rapid emergence in pathogens. The current antibiotic crisis is an evolutionary phenomenon and mitigation strategies needs to account for microbial ecology. It is the rapid acquisition of resistance by pathogens that were previously sensitive leading to therapy failures that is problematic, especially when very few novel antimicrobials are expected to reach the market 3 .
Mechanisms driving the emergence and dissemination of the resistome
The resistome refers to the complete set of genes that encode for antibiotic resistance (AR)-related proteins or those proteins that could potentially evolve into powerful AR agents 4 (figure 1). This includes recognized AR genes in pathogenic bacteria (the problematic ones), AR genes from antibiotic-producing organisms, such as Streptomyces spp. producing streptomycin antibiotic and associated resistance gene[5], cryptic AR genes (i.e., genes that could provide resistance in a different genetic setting; e.g., upregulated efflux pumps or downregulated porins), and precursor AR genes that code proteins with a minimal level of affinity or resistance to antibiotic compounds.
It is noteworthy that a considerable portion of these gene subsets overlap due sequences homology, indicating they likely have a shared evolutionary background.
Image
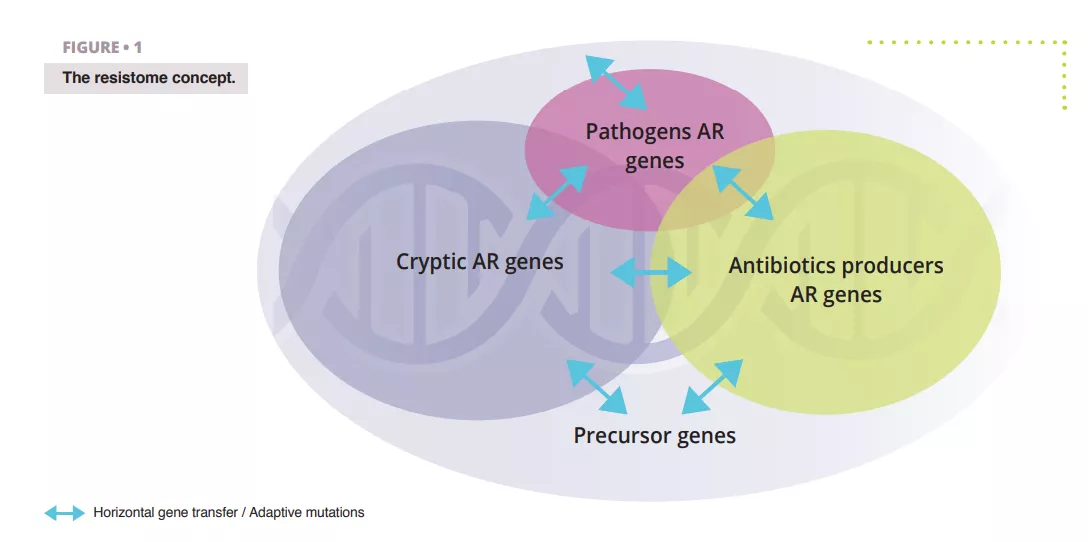
The notion of resistome is distinct from “functional and clinically relevant AR” important. Indeed, resistome genes can transition between the different states described above by horizontal gene transfer (HGT), point mutations, or recombination which are leading to new hosts or genetic context where clinically relevant AR phenotype can be expressed. A resistance gene is therefore not problematic per se as this is host and genetic context dependent, but all resistome genes are potential threat with different associated public health risk outcomes. The discovery of a resistance gene towards a clinically relevant molecule, located on a mobile element and hosted by a human pathogen is a critical risk but the same gene, or close homologues, found in a non-pathogenic soil bacterium and not associated with mobile genetic element is a very low risk ARG. Ranking risk of antibiotic resistance in resistome studies is therefore of paramount.
Horizontal gene transfer (HGT) is a key mechanism responsible for the rapid spread of AR genes among bacteria, even across distant lineages. For example, Bacteroides spp., a predominant group in the human gut microbiota, possess macrolide resistance gene ermB identical to those found in several Clostridium perfringens, Streptococcus pneumoniae and Enterococcus faecalis isolates from various geographical origin, indicating genetic connection between Bacteroides and some Gram-positive bacteria that are not prevalent in the human gut 6 . Genetic elements, such as plasmids, facilitate the transfer of resistance genes between different microbial species 7 . HGT enables the dissemination of genes across diverse environments and bacterial populations, contributing to the overall prevalence and diversity of AR. Co-selection is another significant factor in the spread of AR. The use of non-antibiotic compounds, such as heavy metals and biocides, can co-select for AR genes by exerting selective pressures on microbial populations, either by co-resistance (different resistance determinants present on the same genetic element) and cross-resistance (the same genetic determinant responsible for resistance to antibiotics and metals) 8 . Exposure to naturally occurring antimicrobial compounds such as those produced by competing microorganisms or any co-selective compounds can drive the selection of resistant strains 9 . The presence of antibiotics in the environment, either from natural sources or human activities, further contributes to the selection pressure for resistance. Additionally, the use of antibiotics in agriculture and veterinary practices can lead to the contamination of the environment, promoting the emergence and spread of antibiotic environmental resistance genes overtime 10 .
Understanding barriers and trends to the transfers of genetic vectors harboring resistance genes is crucial to devise eco-evolutionary informed mitigations strategies to limit the dissemination of AR in the early life and in general in clinical settings.
Dysbiosis and the gut resistome in infants: A delicate balance
The environmental reservoir diversity of AR genes and its potential to be transfered poses a threat to the early life human gut microbiome. Strategies such as improved wastewater treatment, responsible use of antibiotics in agriculture and veterinary medicine, and reducing environmental contamination with antibiotic residues and antibiotic resistant bacteria can help mitigate the spread of resistance 11 . Additionally, monitoring and surveillance of environmental reservoirs can provide valuable insights into the emergence and persistence of AR and inform public health interventions.
Adopting a One Health approach, considering the interconnectedness of human health, animal health, and the environment, is essential for addressing the complex challenges posed by AR.
Image
Our gut is rapidly colonized after birth by microorganisms acquired from their mothers and their surrounding environment. It is during the first years of life that changes are drastic and characterized by a low resilience compared to the more stable healthy adult’s gut microbiome. Newborns and infants are therefore more prone to disruptions in microbial communities, known as dysbiosis. During that period, many factors can influence and perturbate gut maturation and potentially lead to long-term consequence on health 12 . Mice studies showed that during this critical developmental window, rather than a direct effect of antibiotic molecules, it is the alteration of the gut microbiota composition that is triggering metabolic consequences, such as obesity 13 .
Unraveling the antibiotic resistome in infants gut: Insights from a large cohort study
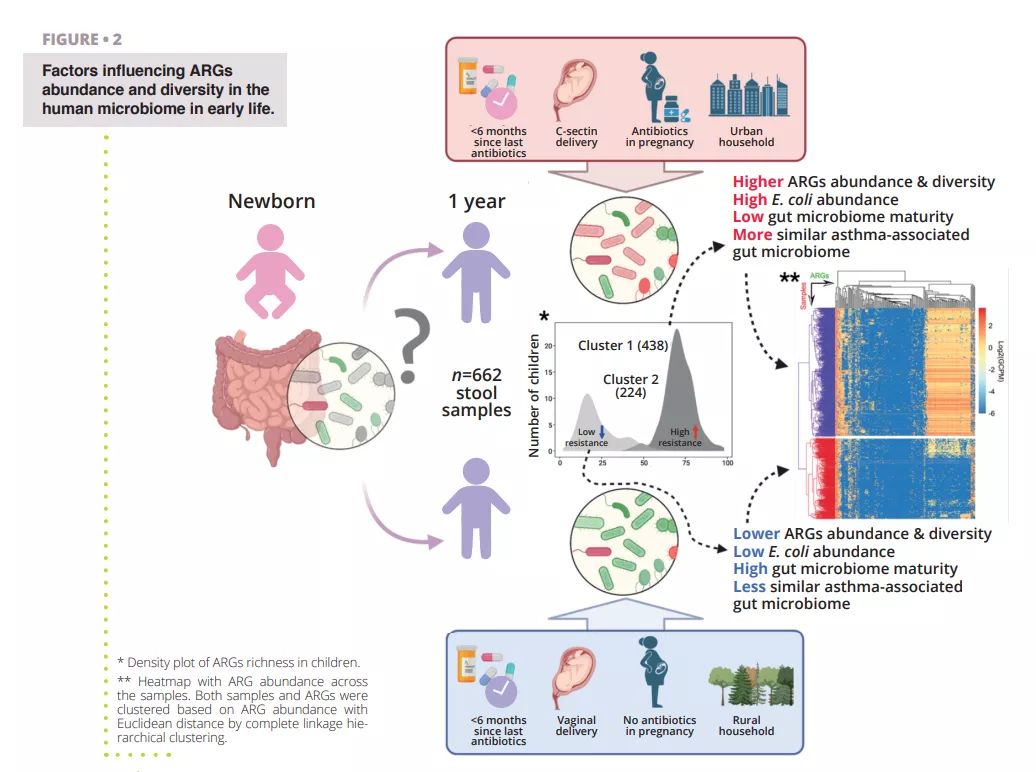
While antibiotic resistance is problematic at all age of life, the establishment of the gut microbiome at early age represent a window of opportunity to limit the buildup of an AR genes reservoir in the gut. It is important therefore to identify the various factors increasing or reducing the abundance of AR genes that could spread to infectious pathogens and cause antibiotic therapy failure throughout life. To study the global human gut resistome researchers rely on holistic approaches interrogating both species presence and genomes functional potential, including the antibiotic resistome. Researchers rely on environmental DNA extraction from proxy samples (e.g., stool samples for the gut) followed by untargeted high-throughput sequencing (shotgun metagenomics). Approximately 80% of human gut bacterial species detected by molecular tools are unculturable, especially the specialized anaerobes inhabiting the gut. It is likely that many microorganisms are organized in multi-species cell aggregates with metabolic co-dependencies making pure strain isolation delicate if not impossible. Using computational methods, it is however possible to reconstruct quasi-complete genomes from metagenomes and associate encoded genes with specific species or even strains of bacteria. In a recent study, researchers analyzed fecal samples from 662 infants from a cohort which followed children from birth to age 7 14 . The objectives of the study were to establish a cohort scale overview of the resistome at 1 year of age and identify perinatal and environmental factors associated with ARG abundance and diversity. The researchers used shotgun metagenomic sequencing of samples obtained at 1 year of age from the 662 children to identify ARGs and bacterial taxa present in the samples (figure 2). Making use of their large dataset, the authors were able to reconstruct Metagenomes Associated Genomes (MAGs), allowing them to confidently annotate the taxonomy of recovered genomes and their AR genes content.
A first result observed was that all children had at least one type of multiple drug ARG in their gut, indicating that even in absence of antibiotic treatment, there is a resident resistome associated with the gut microbiome. Indeed, many multidrug resistance genes were identified as efflux-pumps. These proteins are normal component of every bacterial cell but some can confer AR and are very easily co-selected by cross-resistance, for example towards heavy metals or biocides, potentially explaining their high abundance in the gut, but also in all environments 8, 15 . Another striking result was the clear separation of the cohort in two groups based on their resistome profile. The first group was characterized by a higher ARGs diversity and relative abundance with Escherichia coli as the main contributor of ARGs, as shown in (figure 2). This agrees with previous observations that Enterobacteriaceae are abundant early in life but should decrease rapidly when Bacteroidetes population starts colonizing the gut. Alteration of this maturation in some children can be associated to a combination of several factors, such as antibiotic usage, mode of delivery, rural or urban household that seems to delay the reduction of the population of Escherichia coli and lead to an increased resistome at 1 year of age. This is also confirmed by the observation that the higher abundance of ARGs is associated with a lower gut microbiome maturity score, based on ratios of specific age-related taxa 16 .
Antibiotic treatment during pregnancy and the first year of life of course had significant impact on the abundance of the infant gut resistome. A clear temporal trend could be observed between the time (more than 6 months or less) since the last antibiotic course taken by a child and the abundance of ARGs in its gut.
This indicates some level of resilience at an early age which could be potentially improved with targeted intervention, e.g., with pro- or prebiotics, and remains to be tested. At the cohort scale, the largest differences in ARG abundance were however found for resistance genes towards antibiotic that were not prescribed to the children, underlying that perinatal and environmental factors besides antibiotic therapy are also driving the gut resistome. Another observation from that study that connects the surrounding environment, and associated resistome, and the gut resistome was that children whose households was in urban areas had a significantly higher load of ARGs than children living in rural areas. There is a myriad of potential confounding factors that could explain this but it strengthens the fact that the environment contribution to the development of the early life microbiome is extremely important.
It can be hypothesized that urban living is associated with less contact with the outdoor and a decreased microbiome diversity, or that the type of lodging found in rural (house) or urban (appartement) environment are associated have consequences on the indoor microbiome as seen in bed dust.
Conclusion
Antibiotic resistance is ancient and prevalent and the environmental resistome is a reservoir of potential clinical resistance genes. Mechanisms such as horizontal gene transfer, intricate population dynamics, and co-selection effects, have been identified as significant accelerators in the evolution and spread of the antibiotic resistome making previously sensitive strains of pathogens now resistant to antibiotic therapy. The human gut microbiome and its early developmental phase is prone to dysbiosis and potential invasion, selection, and co-selection of bacteria harboring resistance genes, which can have long term health consequences. It is unclear to what extent these are related to future failed antibiotic therapies. High temporal resolution, long term multi-omics studies are needed to identify first longitudinal patterns of gut microbiome development and second, most probable routes of AR genes transfer by HGT. Together, these results will provide insights to devise preventive strategies for minimizing the development of AR already early in life and limit transfer to problematic pathogens. This effort is of the utmost importance to reduce the burden of the worldwide AMR pandemic and improve global public health by preserving the efficacy of the few antibiotic available to fight infectious diseases.
Sources
1. Nesme J, Cécillon S, Delmont TO, et al. Large-scale metagenomic-based study of antibiotic resistance in the environment. Curr Biol 2014; 24: 1096-100.
2. D’Costa VM, King CE, Kalan L, et al. Antibiotic resistance is ancient. Nature 2011; 477: 457-61.
3. Renwick MJ, Brogan DM, Mossialos E. A systematic review and critical assessment of incentive strategies for discovery and development of novel antibiotics. J Antibiot 2016; 69: 73-88.
4. Wright GD. The antibiotic resistome: the nexus of chemical and genetic diversity. Nat Rev Microbiol 2007; 5: 175-86.
5. Benveniste R, Davies J. Aminoglycoside antibiotic-inactivating enzymes in actinomycetes similar to those present in clinical isolates of antibiotic-resistant bacteria. Proc Natl Acad Sci USA 1973; 70: 2276-80.
6. Shoemaker NB, Vlamakis H, Hayes K, et al. Evidence for extensive resistance gene transfer among Bacteroides spp. and among Bacteroides and other genera in the human colon. Appl Environ Microbiol 2001; 67: 561-8.
7. Martínez JL, Coque TM, Baquero F. Prioritizing risks of antibiotic resistance genes in all metagenomes. Nat Rev Microbiol 2015; 13: 396.
8. Baker-Austin C, Wright MS, Stepanauskas R. Co-selection of antibiotic and metal resistance. Trends Microbiol 2006; 14: 176-82.
9. Wencewicz TA. Crossroads of Antibiotic Resistance and Biosynthesis. J Mol Biol 2019; 431: 3370-99.
10. Knapp CW, Dolfing J, Ehlert PA, et al. Evidence of increasing antibiotic resistance gene abundances in archived soils since 1940. Environ Sci Technol 2010; 44: 580-7.
11. Wolters B, Hauschild K, Blau K, et al. Biosolids for safe land application: does wastewater treatment plant size matters when considering antibiotics, pollutants, microbiome, mobile genetic elements and associated resistance genes? Environ Microbiol 2022; 24: 1573-89.
12. Dogra S, Sakwinska O, Soh SE, et al. Rate of establishing the gut microbiota in infancy has consequences for future health. Gut Microbes 2015; 6: 321-5.
13. Cox LM, Yamanishi S, Sohn J, et al. Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell 2014; 158: 705-21.
14. Li X, Stokholm J, Brejnrod A, et al. The infant gut resistome associates with E. coli, environmental exposures, gut microbiome maturity, and asthma-associated bacterial composition. Cell Host Microbe 2021; 29: 975-87.e4.
15. Mulder I, Siemens J, Sentek V, et al. Quaternary ammonium compounds in soil: implications for antibiotic resistance development. Rev Environ Sci Bio/Technology 2018; 17: 159-85.
16. Subramanian S, Huq S, Yatsunenko T, et al. Persistent gut microbiota immaturity in malnourished Bangladeshi children. Nature 2014; 510: 417-21.